Targeted DNA methylation sequencing
From epigenetics to evolutionary conservation, many of today’s methylation sequencing needs are complex, time-intensive, and potentially costly. Partner with xGen NGS to accelerate your methyl sequencing projects with enhanced data outputs.
xGen™ NGS— made to differentiate.
Overview
- Tried and true products—curated for targeted methylation sequencing
- Library preparation from low input—input as low as 5 ng†
- Powered by xGen Adaptase™ technology—template independent adapter attachment reduces workflow time while generating a less biased library
- Cost effective and reliable—key targets are enriched and sequenced deeply
- Comprehensive sample representation—uniform methylome genome coverage by targeted bisulfite sequencing
- Customizable—select between a small, focused panel or a large panel depending on your research needs
- Workflow design for easy automation
† Before bisulfate conversion.
What is targeted methyl-seq?
Methylation sequencing (methyl-seq), also referred to as bisulfite sequencing, is a next generation sequencing (NGS) approach that researchers use to study methylation patterns of DNA. These patterns can be used to identify different cell types in epigenetics and oncology research [1,2]. Researchers can study the methylation status of the entire genome using whole genome bisulfite sequencing (WGBS) or target specific regions of the genome using hybridization capture panels to enrich genomic regions of interest. More specifically, a targeted methyl-seq approach (also called targeted bisulfite sequencing) uses hybridization probes to capture and enrich biologically relevant regions of interest but with deeper sequencing depth and higher coverage than WGBS.
Targeted methyl-seq workflow
A targeted methyl-seq workflow provides researchers with a streamlined procedure for targeted methylation sequencing. The xGen Methyl-Seq DNA Library Prep Kit has a post-bisulfite library preparation workflow to maximize library complexity by converting bisulfite-induced single-stranded DNA fragments directly into library molecules. This kit is compatible with a range of input quantities and libraries can be generated in as little as 2 hours from bisulfite‑converted samples.
The addition of an xGen Custom Hyb Panel to this workflow enables researchers to capture biologically relevant regions of the genome for targeted methyl-seq. When used for targeted methyl-seq, the xGen Custom Hyb Panel results in sequences with a high on-target mapping percentage and efficiency (Figure 1), as well as methylation profiles that are highly correlated to those obtained from WGBS (Figure 2). Further details on the xGen Custom Hyb Panel for targeted methyl-seq can be found in the AppNote, Cost-effective targeted methyl-seq using an xGen Custom Hyb Capture Panel and the xGen Methyl-Seq DNA Library Prep Kit identifies DNA methylation in low input samples consistent with WGBS.
The recommended group of xGen methyl-seq product offerings also include the compatible core reagents the xGen Hybridization and Wash v2 Kit, xGen Universal Blockers TS, and xGen Library Amplification Primer Mix to accomplish methyl-seq with an easy and flexible workflow.
Together, these xGen NGS products offer you a comprehensive workflow for accelerating your epigenetic research using a targeted methylation sequencing approach.
Transform Your NGS Workflow with Automation
Looking to streamline your NGS workflows? Discover how automation can enhance efficiency and consistency in your lab with our NGS Automation solutions.
Extraction
Bisulfite conversion
Library prep
xGen Methyl-Seq DNA Library Prep Kit (96 rxn)
Converts bisulfite converted fragments into ready-to-sequence libraries in a ~2-hour workflow. Input 1–100 ng for library prep in a targeted methyl-seq workflow†
xGen UDI Primer Plate 1, 8 nt
† Before bisulfate conversion.
Sequencing & analysis
Method data
xGen targeted methyl-seq workflow results in comprehensive coverage with low-input samples
High-quality sequencing results with the xGen Methyl-Seq DNA Library Prep Kit plus xGen Custom Hyb Panel workflow
To assess the xGen Methyl-Seq DNA Library Prep Kit plus xGen Custom Hyb Panel workflow, libraries were prepared and enriched from 5, 10, or 25 ng of cell-free DNA (cfDNA) reference material (RM) (Horizon HD833). The methylation‑specific xGen Custom Hyb Panel was designed to enrich for five genes associated with methylation biomarkers, covering a total region of 128 kb. Hybridization capture was performed on all libraries following the xGen Hyb and Wash Reagents v3 Kit protocol, hybridizing three libraries per capture for 16 hours. Sequencing was performed on a NextSeq™ 500 (Illumina®) platform using 2 x 75 bp reads with 10% PhiX spike-in. The results of this workflow were assessed by generating common sequencing quality metrics outlined in Figure 1 using the Picard toolkit [3].
The xGen Methyl-Seq DNA Library Prep Kit, provides researchers with flexibility as it can prepare libraries from a wide range of sample inputs, which can then be enriched with a specially designed xGen Custom Hyb Panel to obtain high‑quality sequences from regions of interest. Together, the xGen Methyl-Seq Library Prep Kit and an xGen Custom Hyb Panel create a meaningful tool for accelerating focused epigenetic research.
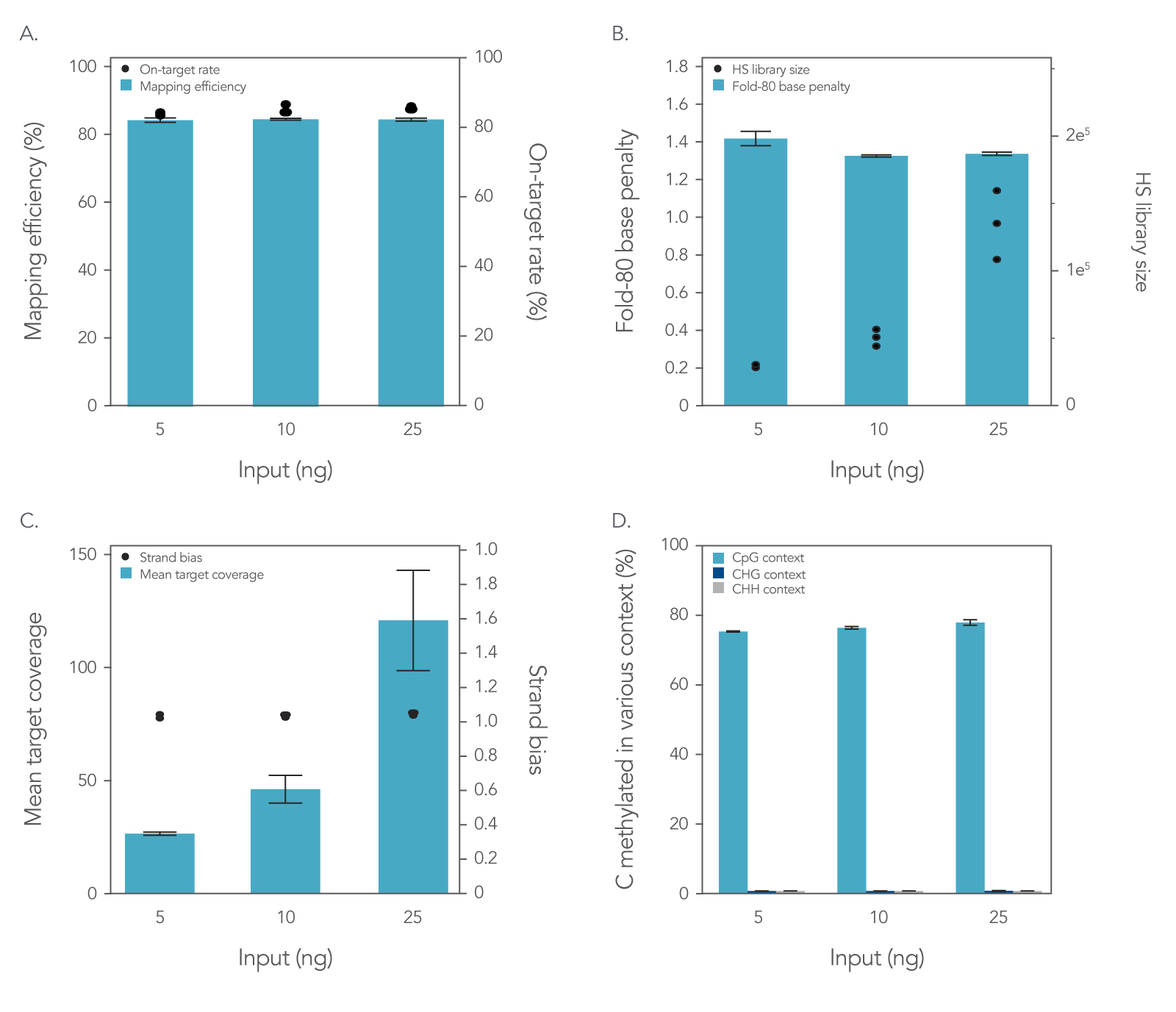
Figure 1. Libraries enriched with the xGen Custom Hyb Panel for targeted methyl-seq result in high-quality sequencing results. Hybridization capture results using a 128 kb xGen Custom Hyb Panel with libraries made from 5 ng (n = 2), 10 ng (n = 3), and 25 ng (n = 3) of cfDNA RM were sequenced on a NextSeq™ 500 (Illumina) instrument and subsampled to 2 million reads per sample. The data showed equivalently (A) high on-target percentages and high mapping efficiency and (B) fold-80 scores close to 1. As expected, hybrid-selection (HS) library size (B) and (C) the mean coverage depth increased when higher amounts of input DNA were used. (D) A similar percentage of C’s methylated in various contexts was seen across the target space for all sample inputs. Post-capture libraries from these figures were amplified using KAPA 2x HiFi PCR Mix, as these data were generated prior to release of xGen 2x HiFi PCR Mix. Error bars represent the standard deviation of replicates.
Reliable methylation profiles from libraries enriched with the xGen Custom Hyb Panel
To determine if the xGen Custom Hyb Panel preserved methylation signatures, the methylation percent per CpG site for a whole-genome bisulfite sequencing (WGBS) library prepared using 10 ng of cfDNA was compared to the same metric obtained from the 10 ng cfDNA library enriched with the xGen Custom Hyb Panel. Across shared target spaces, the xGen Custom Hyb Panel and the WGBS library were highly correlated (Pearson, r ≥ 0.97; Figure 2).
The high correlation between libraries enriched using a methyl-specific xGen Custom Hyb Panel and the libraries obtained from WGBS shows that the xGen Custom Hyb Panel is a reliable, cost-effective way to assess methylation of key targets associated with different variants.
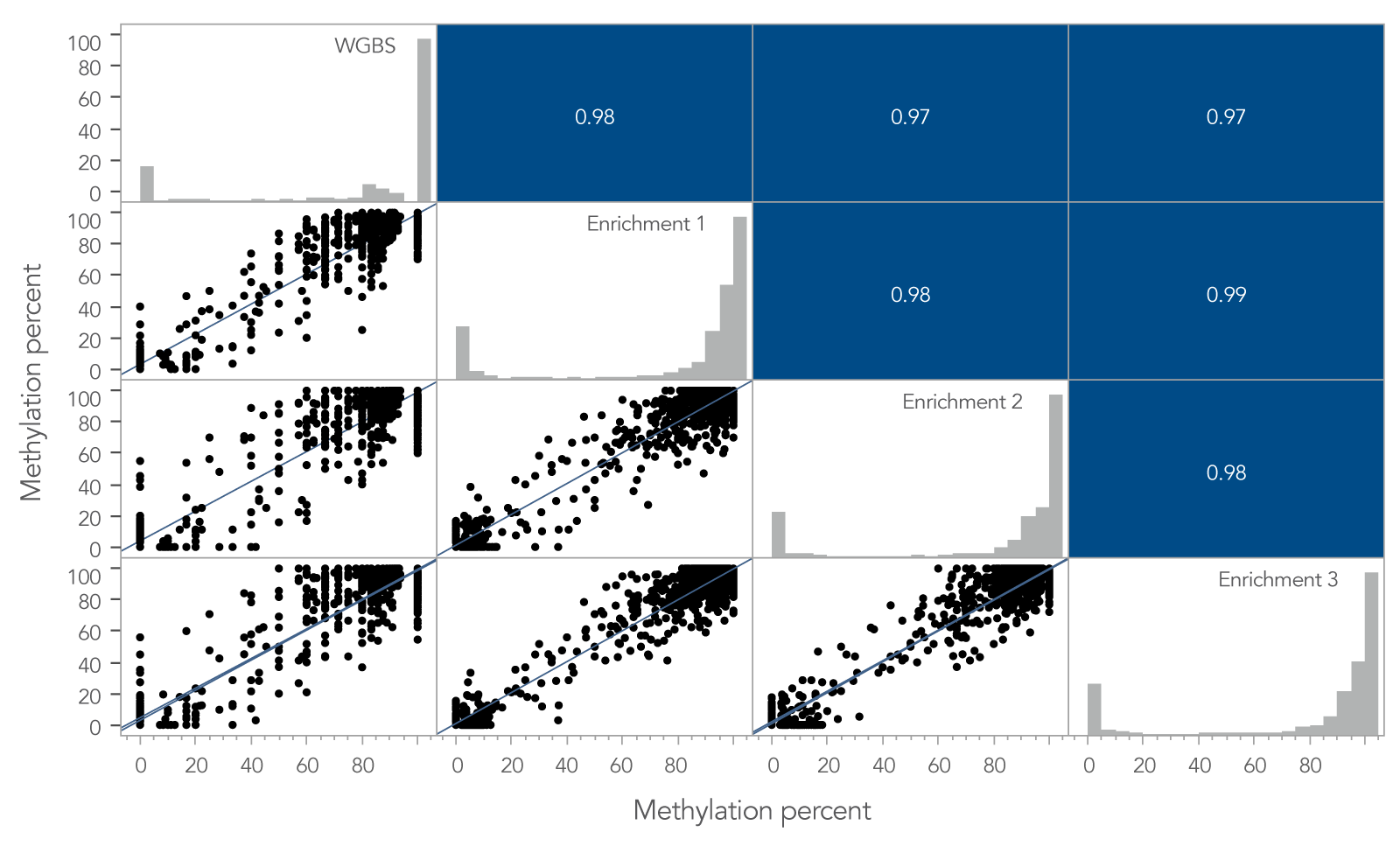
Figure 2. Methylation profiles of targeted methyl-seq libraries and WGBS libraries are highly correlated. Pearson’s correlation between target enriched xGen Methyl-Seq libraries and libraries from WGBS. Per-CpG site methylation for three captured libraries and one whole‑genome library made from 10 ng cfDNA reference material. In total, 860 M reads for WGBS and 2 M reads for samples enriched using a methyl-specific xGen Custom Hyb Panel were used to compare the methylation percent per CpG site for any site that had ≥ 5X coverage in the shared target space. A high correlation of site-specific methylation is observed between whole‑genome and target enriched libraries, including regions containing hypo-, hyper- and intermediate methylation. Post-capture libraries from these figures were amplified using KAPA 2x HiFi PCR Mix, as these data were generated prior to release of xGen 2x HiFi PCR Mix.
Ordering
Use our xGen Hyb Panel Design Tool to create your xGen custom Hyb panels.
Resources
References
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484-492.
- Moarii M, Boeva V, Vert JP, et al. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genomics. 2015;16:873.
- “Picard Toolkit.” Broad Institute, GitHub repository: Broad Institute; 2019